Methods
Bioinformatics and statistical analysis were run in R software v.3.5.3 [1]
1. Systematic review and selection of studies
A systematic search of studies published in the period 2011-2019 was conducted in October 2019 following This review was conducted in October 2019, according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement guidelines [52]. We searched breast cancer methylation data studies in GEO database [53], using the keywords: “breast cancer”, “methylation” and “Homo sapiens" in studies published in English. We applied the following exclusion criteria: (i) studies conducted in organisms other than humans; (ii) sample size less than 10 in each experimental group; (iii) different methylation profiling platform to Infinium HumanMethylation450 BeadChip from Illumina and (iv) experimental design different from case-control.
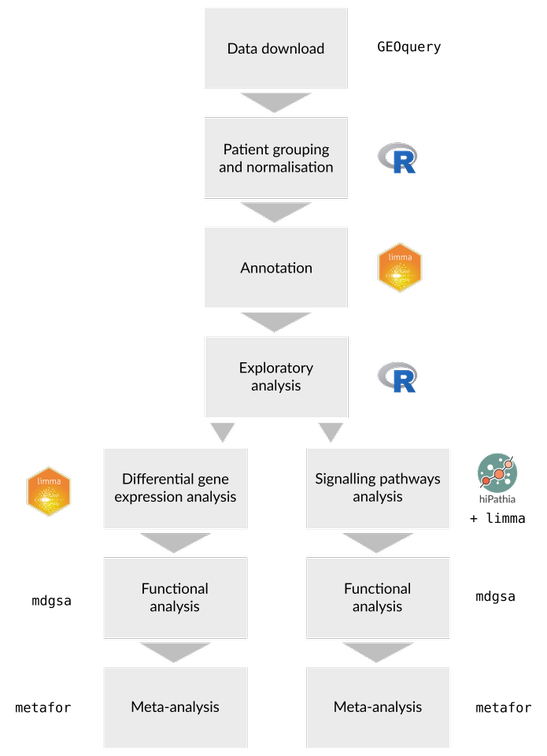
2. Bioinformatics analysis strategy
The following procedure was applied to each of the individually selected studies: i) Data acquisition. ii) Exploratory analysis and quality control of the samples. iii) Then, the studies were separated into different "case vs. control" comparisons, i.e. if a study consists of healthy samples and samples from two breast cancer subtypes we will obtain two comparisons —healthy samples vs. breast cancer subtype I and healthy samples vs. breast cancer subtype II—. iv) Analysis of differential methylation between case and control groups by comparison. v) Analysis of functional enrichment for each of the comparisons. vi) Integration of the methylation profiles and functional results in the final meta-analysis
3. Data exploration, quality control and normalization
Exploration of raw data was performed through principal component analysis (PCA) and clustering analysis. Data quality control was conducted with minfi [24] R package. In this step we check levels of the signal from the methylated and non-methylated channels, then a quantile normalization was performed on the data to compare all together
4. Individual epigenomic analysis
The differential methylation analysis for each of the different comparisons was carried out with the minfi R package. For each comparison, permutation tests were performed to obtain the methylation scores of all analyzed regions on the Illumina BeadChip 450k. A total of 7 comparisons derived from the individual studies were performed: Control vs TNBC, Control vs HER-2, Control vs Lum-A, Control vs Lum-B, Control vs Brcmutant, Control vs Invasive Ductal Carcinoma —IDC— and Control vs Invasive Lobular Carcinoma —ILC—.
Subsequently, methylation scores were annotated to gene level through the bumphunter R package [54]. In cases where more than one differentially methylated region (DMR) existed for a gene, the one with the highest absolute value of differential methylation was used. Then, a functional enrichment analysis was performed from the differential methylation results using Gene Set Analysis (GSA) [55]. To do this, the genes were ordered according to their p-values and the sign of the contrast statistic. The GSA was then performed following the logistic regression model implemented in the mdgsa R package [56], as well as its corresponding functional annotation. The p-values were corrected for each function for false discovery rate (FDR) [57]. The databases used for functional enrichment were the Gene Ontology [58] (GO) and the Kyoto PATHWAY Encyclopedia of Genes and Genomes [59] (KEGG).
Significant functions have been represented in the form of Upset plots [60]. With this type of graph we can see the number of functional elements specific and shared by each of the different breast cancer subtypes studied
5. Methylated genes meta-analysis
From the mapping of the differentially methylated regions, the genes included in them were identified. In order to obtain an integrated level of methylation in all the studies, those genes with a common differential pattern between the case and control groups were selected. For each gene, the p-values of all comparisons were then combined using the Fisher combination method (or the inverse normal/weighted method). This meta-analysis strategy provided the genes with a significant common methylation profile in all the selected studies
6. Functional DNA methylation signatures meta-analysis
Finally, once the functional enrichment study was performed for each of the individual comparisons the results were integrated into a functional meta-analysis. The methodology of the functional meta-analysis followed was the one proposed by [17]. For this task, the metafor [61] R package was used to assess the combined effect of the studies together with a random effects model. The results of the meta-analysis are more robust than the results of individual comparisons due largely to the larger sample size. The variability of individual studies was taken into account in the meta-analysis so that if a study had little variability, it would carry more weight in the meta-analysis and be more influential in the calculation of the log odds ratio (LOR). In turn, both a heterogeneity analysis to check the suitability of the different studies selected and a sensitivity analysis and assessment of bias to detect whether any of the comparisons had an excessive influence on the final meta-analysis were performed.
Each function analysed in the meta-analysis is accompanied by the combined estimate of the effect of the studies (LOR), the 95% confidence interval and the p-value adjusted by the Benjamini and Hochberg method. Thus, those functions with an adjusted p-value equal to or less than 0.05 were considered significant. For each significant function, forest and funnel plots were used to measure the contribution of each study to the meta-analysis and to assess its variability
References
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009;6:e1000097. Available from: https://dx.plos.org/10.1371/journal.pmed.1000097
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012;41:D991-D995. Available from: http://academic.oup.com/nar/article/41/D1/D991/1067995/NCBI-GEO-archive-for-functional-genomics-data
- Aller R, Fernández-Rodríguez C, lo Iacono O, Bañares R, Abad J, Carrión JA, et al. Consensus document. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol. y Hepatol. English Ed. 2018;41:328-349. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0210570518300037
- R Core Team. R: A Language and Environment for Statistical Computing. 2019;Available from: http://www.r-project.org/
- Maglott D. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2004;33:D54-D58. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gki031
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185-193. Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/19.2.185
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. Available from: http://genomebiology.biomedcentral.com/articles/10.1186/gb-2010-11-3-r25
- Davis SR, Lambrinoudaki I, Lumsden M, Mishra GD, Pal L, Rees M, et al. Menopause. Nat. Rev. Dis. Prim. 2015;1:15004. Available from: http://www.nature.com/articles/nrdp20154
- Freeman EW, Sammel MD, Sanders RJ. Risk of long-term hot flashes after natural menopause. Menopause. 2014;21:924-932. Available from: http://journals.lww.com/00042192-201409000-00005
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47-e47. Available from: https://doi.org/10.1093/nar/gkv007
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-140. Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btp616
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B. 1995;57:289-300. Available from: http://www.jstor.org/stable/2346101
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 2005;102:15545-15550. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0506580102
- Montaner D, Dopazo J. Multidimensional Gene Set Analysis of Genomic Data. PLoS One. 2010;5:e10348. Available from: https://dx.plos.org/10.1371/journal.pone.0010348
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25-29. Available from: http://www.nature.com/doifinder/10.1038/75556
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27-30. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/27.1.29
- Lex A, Gehlenborg N. Sets and intersections. Nat. Methods. 2014;11:779-779. Available from: http://www.nature.com/articles/nmeth.3033
- Hidalgo MR, Cubuk C, Amadoz A, Salavert F, Carbonell-Caballero J, Dopazo J. High throughput estimation of functional cell activities reveals disease mechanisms and predicts relevant clinical outcomes. Oncotarget. 2017;8. Available from: http://www.oncotarget.com/fulltext/14107
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res.. 2016;45:D158-D169. Available from: https://doi.org/10.1093/nar/gkw1099
- García-García F. Métodos de análisis de enriquecimiento funcional en estudios genómicos. 2016. Available from: https://www.educacion.gob.es/teseo/mostrarRef.do?ref=1307283
- Viechtbauer W. Conducting Meta-Analyses in R with the metafor Package. J. Stat. Softw. 2010;36. Available from: http://www.jstatsoft.org/v36/i03/